Chapter 8 Plotting taxonomy
8.1 Introduction
In this chapter you will learn how to plot a custom taxa barplot to compare relative abundances between different sets of samples or treatments.
8.1.1 Required files or objects
- A
phyloseqobject; either read in from a pre-saved.rdsfile or created as described in chapter 6 under “Import qiime-files and create a phyloseq object”.
- If you dont have your own data you can download the pre-saved
.rdsobject from Chapter 6 and follow the below steps: ps_ProjectX_2022July. This object is based on publicly available data from anaerobic sludge of Danish wastewater treatment plants.
8.2 Workflow
8.2.2 Load phyloseq object
Do this if you saved a phyloseq object in .rds format before. Otherwise create a phyloseq object as described in chapter 6 under “Import qiime-files and create a phyloseq object”.
# reading in a previously saved phyloseq object
ps <- readRDS('ps_ProjectX_2022July')
#ps (not run) to get an overview of number of taxa and samples contained in the phyloseq object
# output
# phyloseq-class experiment-level object
# otu_table() OTU Table: [ 4218 taxa and 51 samples ]
# sample_data() Sample Data: [ 51 samples by 15 sample variables ]
# tax_table() Taxonomy Table: [ 4218 taxa by 7 taxonomic ranks ]
# phy_tree() Phylogenetic Tree: [ 4218 tips and 4217 internal nodes ]8.2.3 Check metadata contained in the phyloseq object.
Now lets check which columns in the metadata contain factors. Factors enable you to determine the order of categorical data, such as treatments, in the ggplots. Learn more about factors here: https://swcarpentry.github.io/r-novice-inflammation/12-supp-factors/index.html.
In this case, the columns Reactor and Location contain factors.
# Use the 'structure' (str) command to get an overview of your data columns
# Learn what are characters, integers or factors,
str(data.frame(sample_data(ps)))## 'data.frame': 51 obs. of 15 variables:
## $ Assay.Type : chr "AMPLICON" "AMPLICON" "AMPLICON" "AMPLICON" ...
## $ Bases : int 13254234 24196788 15758554 20048406 37634632 43555904 35107436 13888140 23172184 35065898 ...
## $ BioProject : chr "PRJNA645373" "PRJNA645373" "PRJNA645373" "PRJNA645373" ...
## $ Bytes : int 9235913 16404070 10758456 13009806 24207581 27963491 22585685 9388862 15125835 22520074 ...
## $ Organism : chr "anaerobic digester metagenome" "anaerobic digester metagenome" "anaerobic digester metagenome" "anaerobic digester metagenome" ...
## $ collection_date: chr "30/11/16" "9/3/16" "17/3/16" "5/12/16" ...
## $ loc : chr "Ejby Moelle" "Soeholt" "Esbjerg Vest" "Viborg" ...
## $ Reactor : Factor w/ 10 levels "1A","1B","2B",..: 4 4 6 7 7 5 5 5 4 4 ...
## $ Type : chr NA NA NA "DS" ...
## $ Purpose : chr "reactorfoampotentials" "reactorfoampotentials" "reactorfoampotentials" "foamnofoamcomparison" ...
## $ Experiment : chr "SRX8715180" "SRX8715169" "SRX8715158" "SRX8715151" ...
## $ lat_lon : chr "55.398077 N 10.415041 E" "56.175302 N 9.582588 E" "55.488235 N 8.430655 E" "56.425367 N 9.4527943 E" ...
## $ ReleaseDate : chr "2021-01-01T00:00:00Z" "2021-01-01T00:00:00Z" "2021-01-01T00:00:00Z" "2021-01-01T00:00:00Z" ...
## $ Sample.Name : chr "16SAMP-17236" "16SAMP-17231" "16SAMP-17233" "MQ170915-14" ...
## $ Location : Factor w/ 12 levels "Avedoere","Damhusaaen",..: 4 11 5 12 12 12 12 2 11 11 ...8.2.4 Colors
Before you create ggplots to compare taxonomic compositions between samples and treatments it is helpful to create a named color vector that will apply to all your plots and ensures that each taxonomic name is a specific color across all your plots.
There are different packages that help to create a names color vector. Here we use the package colorspace.
A handy color cheat sheet is available here: https://www.nceas.ucsb.edu/sites/default/files/2020-04/colorPaletteCheatsheet.pdf
First create a vector of all possible taxonomic names that could be included in your ggplot. For example, if you plan to plot on phylum-level then all phyla names are extracted from the phyloseq object and then those names are given a color code. The colors are defined as hex color codes.
# You can check Hex codes with help of https://htmlcolorcodes.com/
# https://www.nceas.ucsb.edu/sites/default/files/2020-04/colorPaletteCheatsheet.pdf
# For all possible phyla names
phylum_data <- microbiome::aggregate_taxa(ps, "Phylum") # aggregate data to phylum level
# the pacakage 'microbiome' has some handy helper functions such as `aggregate`.
names <- rownames(phylum_data@tax_table) # extract a vector of phylum names using `rownames`
# Package colorspace
library(colorspace)
cols <- colorspace::rainbow_hcl(length(names)) # this creates x number of hex codes. x = `length(names)`.
# in this case there are 48 phyla, hence we get 48 hex codes.
names(cols) <- names # name each hex code with phyla names.
# Option to use the package viridis
#library(viridis)
# cols <- viridis::turbo(length(names), direction = -1)
# names(cols) <- names
# Option to use the package RColorBrewer
# library(RColorBrewer)
# display.brewer.all(length(names)) # check colour themes available in the RColorBrewer package
# cols <- brewer.pal(length(names), 'RdYlBu')
# cols <- rainbow_hcl(length(names))
# names(cols) <- names
cols # check content of the color vector## Acetothermia Acidobacteriota
## "#E495A5" "#E3979D"
## Actinobacteriota Armatimonadota
## "#E19995" "#DE9B8D"
## Bacteroidota Bdellovibrionota
## "#DB9D85" "#D79F7E"
## Caldatribacteriota Caldisericota
## "#D2A277" "#CDA471"
## Campilobacterota Chloroflexi
## "#C7A76C" "#C1A968"
## CK-2C2-2 Cloacimonadota
## "#BAAC65" "#B3AE64"
## Coprothermobacterota Cyanobacteria
## "#ABB065" "#A2B367"
## Deinococcota Dependentiae
## "#99B56B" "#90B66F"
## Desulfobacterota Elusimicrobiota
## "#86B875" "#7CBA7C"
## FCPU426 Fermentibacterota
## "#72BB83" "#67BC8A"
## Fibrobacterota Firmicutes
## "#5CBD92" "#51BE9A"
## Fusobacteriota Gemmatimonadota
## "#47BEA2" "#3FBEA9"
## Hydrogenedentes Hydrothermae
## "#39BEB1" "#37BDB8"
## Latescibacterota Margulisbacteria
## "#3BBCBF" "#42BBC6"
## Marinimicrobia_(SAR406_clade) MBNT15
## "#4CB9CC" "#57B8D1"
## Methylomirabilota Myxococcota
## "#64B5D6" "#70B3DA"
## Nitrospirota Patescibacteria
## "#7DB0DD" "#8AADE0"
## Planctomycetota Proteobacteria
## "#96AAE1" "#A1A7E2"
## SAR324_clade(Marine_group_B) Spirochaetota
## "#ACA4E2" "#B5A1E1"
## Sumerlaeota Sva0485
## "#BE9EDF" "#C69BDC"
## Synergistota Thermotogota
## "#CD99D8" "#D497D4"
## Verrucomicrobiota WPS-2
## "#D995CF" "#DD94C9"
## WS1 WS4
## "#E093C3" "#E293BC"
## Zixibacteria Unknown
## "#E493B4" "#E494AD"8.2.5 Pre-filtering
Some arbitrary quality filtering of individual ASVs. For example, here we choose to remove ASVs that have very few reads (i.e. less than 5 counts). Use the prune_taxa() function from phyloseq to filter taxa or the prune_samples() function to filter selected samples.
ps_flt <- prune_taxa(taxa_sums(ps) >= 5, ps) #minimum reads per feature
#ps_flt
# phyloseq-class experiment-level object
# otu_table() OTU Table: [ 3911 taxa and 51 samples ]
# sample_data() Sample Data: [ 51 samples by 15 sample variables ]
# tax_table() Taxonomy Table: [ 3911 taxa by 7 taxonomic ranks ]
# phy_tree() Phylogenetic Tree: [ 3911 tips and 3910 internal nodes ]
# Use `prune_samples()` If you need to filter data to a subset of samples.
# E.g. choose samples of reactor 1A only
ps_flt_1A <- prune_samples(sample_data(ps_flt)$Reactor == "1A", ps_flt)
# Use `prune_taxa` to remove any ASVs/taxa that are now zero
# (i.e. are not present in reactor 1A).
# In other words we keep only those ASVs/Taxa that are present in the filtered samples.
ps_flt_1A <- prune_taxa(taxa_sums(ps_flt_1A) > 0, ps_flt_1A)
#phyloseq-class experiment-level object
#otu_table() OTU Table: [ 420 taxa and 2 samples ]
#sample_data() Sample Data: [ 2 samples by 15 sample variables ]
#tax_table() Taxonomy Table: [ 420 taxa by 7 taxonomic ranks ]
#phy_tree() Phylogenetic Tree: [ 420 tips and 419 internal nodes ]8.2.6 Analysis and plotting
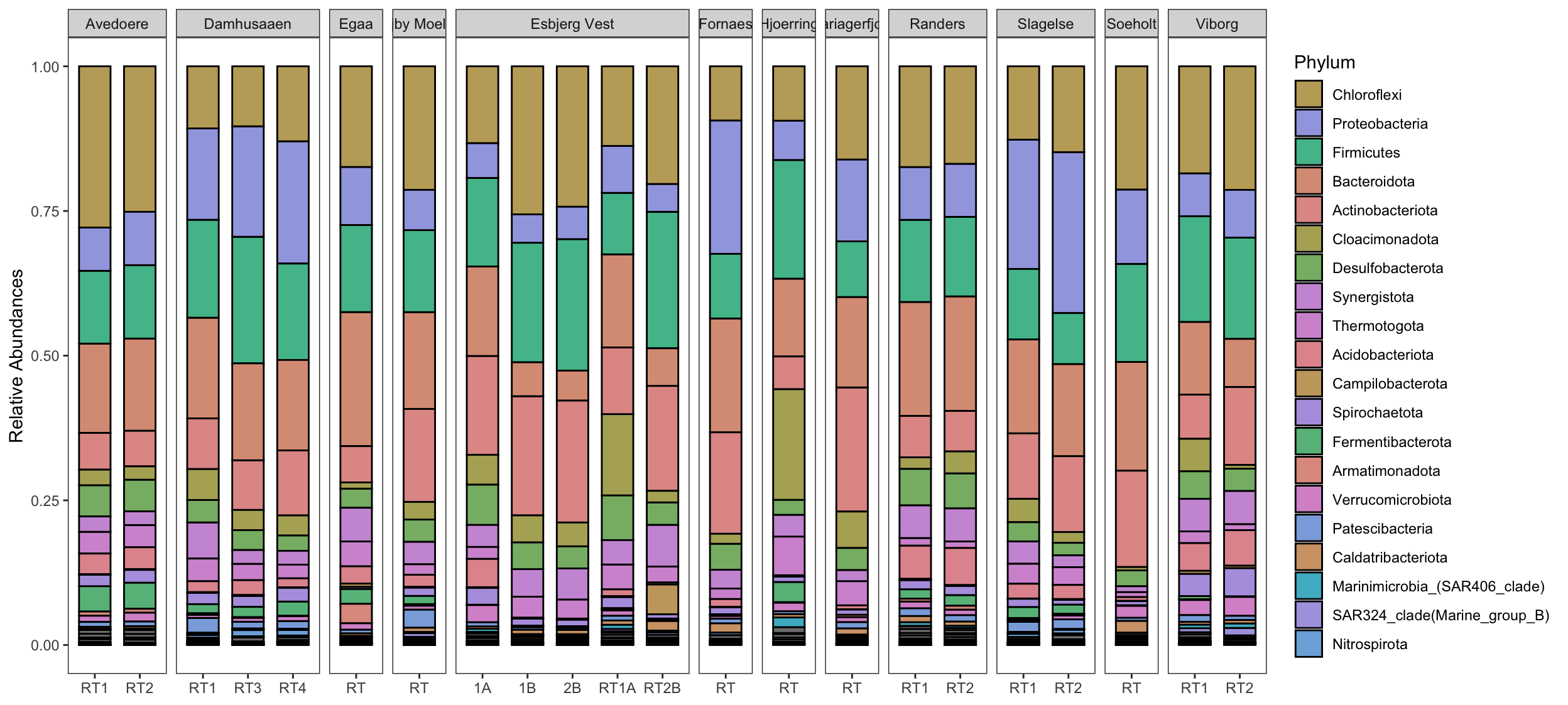
Create stacked barplot comparing relative abundances of the normalised phyloseq object.
First abundances are aggregated to phylum level and then transformed into relative abundances. This is followed by the creation of a long-form abundance table that includes metadata with help of the very handy psmelt function from the phyloseq package. The function psmelt takes a phyloseq object and extracts abundances and related metadata into a dataframe for plotting.
Finally, the abundances of this long abundance table are summarised into the factors that you want to compare. In this example, abundances of different Locations are compared, hence the column Location will be summarised and plotted.
# Phylum plot
# First, create a long abundance data table
taxa_data <- microbiome::aggregate_taxa(ps_flt, "Phylum") %>%
# aggregate abundances to phylum-level
microbiome::transform(., "compositional") %>% # transform abundance to relative abundances
phyloseq::psmelt(.) # extract long abundance table with metadata for plotting.
# Note: The `.` replaces the phyloseq object.
# This is because the functions are piped into a sequence by using the `%>%` operator.
# The `%>%` operator is a Tidyverse function and is very helpful in putting
# various different operations and changes to the data into one sequence.
# We could have put the below operations into the same above sequence too.
# There is no difference in the outcome.
# Summarise abundances by columns of your choice containing factors.
# E.g. compute the mean abundances based on the columns `Location`, `Reactor` and `Phylum.
taxa_data <- taxa_data %>%
dplyr::group_by(Location, Reactor, Phylum) %>% # group by Location and Phylum to summarise data
dplyr::summarise(Meanabu = mean(Abundance)) %>% # Create a column called `Meanabu`
dplyr::filter(Meanabu > 0) %>% # filter out rows that have 0 abundance
dplyr::arrange(desc(Meanabu)) # sort the abundances
# Now, create a vector with phylum names that will determine the order of
# phylum names in the stacked barcharts.
order <- taxa_data$Phylum %>% unique()
# Phylum names need to be factors for plotting.
# This is the final dataframe for plotting
taxa_data$Phylum <- factor(taxa_data$Phylum, levels = order) #
# Check out the first few rows
head(taxa_data)## # A tibble: 6 × 4
## # Groups: Location, Reactor [6]
## Location Reactor Phylum Meanabu
## <fct> <fct> <fct> <dbl>
## 1 Avedoere RT1 Chloroflexi 0.279
## 2 Slagelse RT2 Proteobacteria 0.278
## 3 Esbjerg Vest 1B Chloroflexi 0.256
## 4 Avedoere RT2 Chloroflexi 0.251
## 5 Esbjerg Vest 2B Chloroflexi 0.243
## 6 Esbjerg Vest RT2B Firmicutes 0.236# Finally its time to create a ggplot.
taxaplot <- taxa_data %>%
ggbarplot( x= "Reactor",
y = "Meanabu",
fill = "Phylum",
position = position_stack(),
legend = "right",
ylab = "Relative Abundances") +
# Tell ggplot to use the `cols` vector created earlier
# only the first 20 colors are shown in legend, hence the `order[1:20]`
scale_fill_manual(values = cols,
limits = order[1:20]) +
facet_grid(cols = vars(Location), # create facets by factors in column `Location`.
scales = "free_x",
space = "free_x") +
guides(fill = guide_legend(byrow = FALSE)) +
rotate_x_text(angle = 45) + # rotate the x-axis labels
theme_bw() + # makes ggplots look better
theme(axis.title.x=element_blank(), # remove the background grid
panel.grid.major = element_blank(),
panel.grid.minor = element_blank())
taxaplot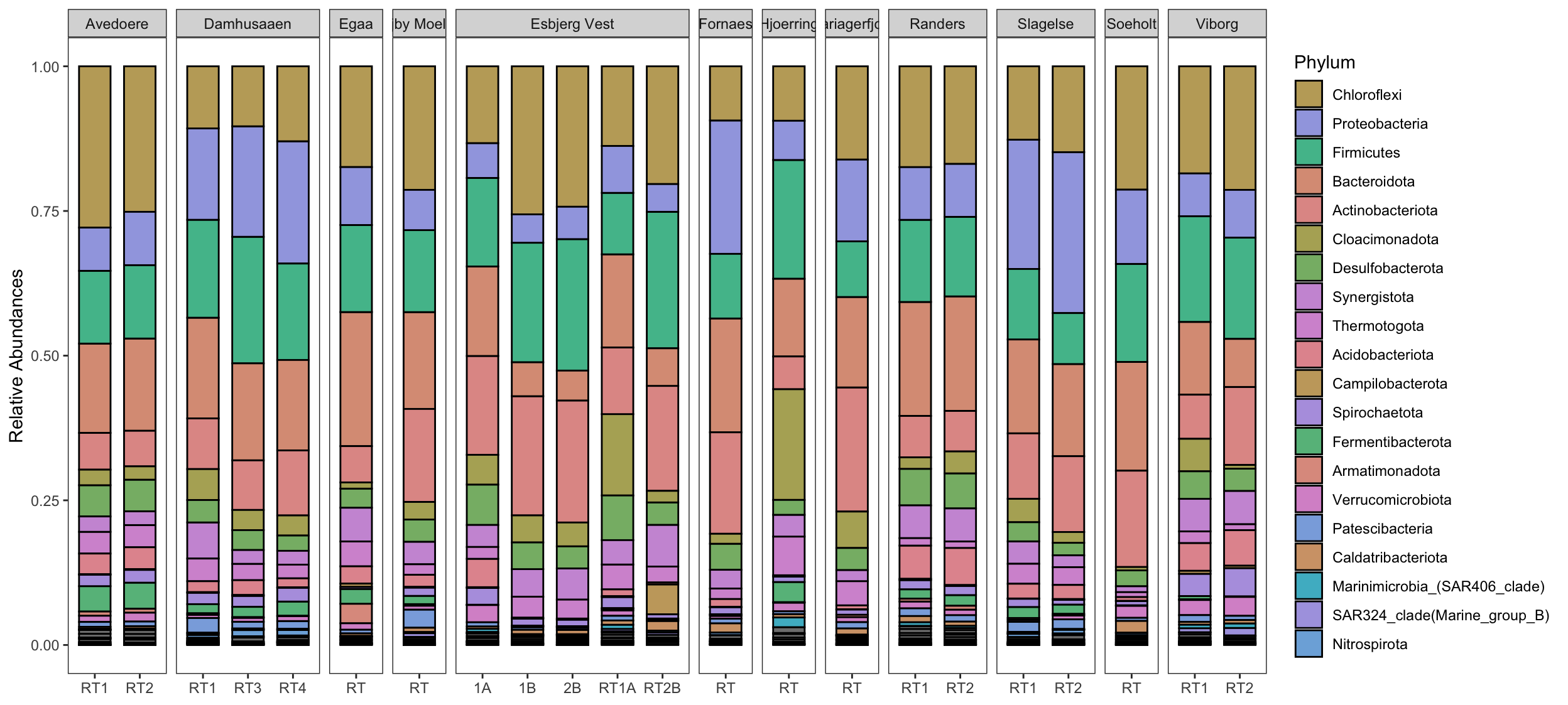
8.2.7 Prevalance table
A prevalence table may assist in getting an overview of the abundances. The mean prevalence is the average number of times that the taxon appears across all samples.
prevalancedf = apply(X = phyloseq::otu_table(ps_flt),
MARGIN = 1,
FUN = function(x){sum(x > 0)})
prevalancedf = data.frame(Prevalence = prevalancedf,
TotalAbundance = phyloseq::taxa_sums(ps_flt),
phyloseq::tax_table(ps_flt))
prevalancedf <- plyr::ddply(prevalancedf, "Phylum", function(df1){
data.frame(mean_prevalence=mean(df1$Prevalence),
total_abundance=sum(df1$TotalAbundance, na.rm = T),
stringsAsFactors = F)
})
# print table from highest to lowest abundance
prevalancedf %>%
arrange(desc(total_abundance)) ## Phylum mean_prevalence total_abundance
## 1 Chloroflexi 5.124069 228197
## 2 Bacteroidota 4.688679 201921
## 3 Firmicutes 4.453435 197683
## 4 Actinobacteriota 5.231343 165740
## 5 Proteobacteria 4.249147 161756
## 6 Desulfobacterota 5.425743 57146
## 7 Synergistota 6.595506 51225
## 8 Cloacimonadota 6.500000 45294
## 9 Acidobacteriota 3.953488 35304
## 10 Thermotogota 10.190476 33489
## 11 Spirochaetota 4.578431 25015
## 12 Verrucomicrobiota 3.382022 16598
## 13 Fermentibacterota 35.000000 14812
## 14 Patescibacteria 3.669173 14399
## 15 Caldatribacteriota 5.500000 8932
## 16 Planctomycetota 2.573171 5455
## 17 Armatimonadota 2.933333 4443
## 18 Marinimicrobia_(SAR406_clade) 20.000000 3818
## 19 <NA> 4.052632 3511
## 20 Hydrogenedentes 3.950000 3411
## 21 Nitrospirota 8.750000 2853
## 22 Campilobacterota 4.187500 2710
## 23 SAR324_clade(Marine_group_B) 4.000000 2616
## 24 WS1 3.625000 2559
## 25 Fibrobacterota 2.826087 1938
## 26 Myxococcota 3.176471 1353
## 27 Bdellovibrionota 3.600000 1111
## 28 Caldisericota 3.818182 1067
## 29 WPS-2 2.666667 897
## 30 Sumerlaeota 3.888889 683
## 31 Cyanobacteria 9.000000 647
## 32 Fusobacteriota 2.888889 565
## 33 Coprothermobacterota 2.800000 511
## 34 Gemmatimonadota 2.875000 300
## 35 Elusimicrobiota 2.833333 225
## 36 CK-2C2-2 4.000000 162
## 37 Methylomirabilota 4.000000 159
## 38 Latescibacterota 2.333333 86
## 39 Dependentiae 2.500000 78
## 40 Zixibacteria 5.000000 74
## 41 Sva0485 2.333333 66
## 42 Acetothermia 2.000000 59
## 43 WS4 1.500000 39
## 44 Hydrothermae 1.000000 26
## 45 Deinococcota 1.000000 118.2.8 Krona charts
On macOS or Linux, a more interactive way to explore the various levels of the taxonomy in your data is to use Krona charts.
All you need is KronaTools and the R package psadd.
To install KronaTools simply download and unzip the repository of https://github.com/marbl/Krona (The green “Code” button) and execute the install.pl file as described in the Installation Wiki at https://github.com/marbl/Krona/wiki/Installing. Once installed you can run the following:
# install a package with custom function for the phyloseq package
# remotes::install_github("cpauvert/psadd")
library(psadd)
# Load ps files
# Use the data-set that we filtered earlier 'ps_flt'
# Separate the output by the metadata column "Location". We will get a krona plot for digesters in each location.
# not run
# plot_krona(ps_flt,"krona-file", "Location",trim=T) # Function plot_krona from the psadd package
# This creates an 'html' file named 'krona-file, which you can open in any web browser. Explore the Krona plot in your web browser: krona-file